

YB∕T 4766-2019 耐火材料用工业硅中单质硅和二氧化硅的测定方法/Chemical analysis of elemental silicon and silicon dioxidein industrial silicon for refractories
耐火材料用工业硅中单质硅和二氧化硅的测定方法
1范围
本标准规定了耐火材料用工业硅中单质硅和二氧化硅的化学分析方法。
本标准适用于耐火材料用工业硅中单质硅和二氧化硅含量的测定,测定范围见表1。
耐火材料中单质硅含量的测定也可参照此标准。
|
表1测定范围 |
|
|
分析项目 |
范围(质量分数)/% |
|
Si |
20~97(稀碱分离法) |
|
0.5~99(气体容量法) |
|
|
SiO2 |
>0.5 |
2 规范性引用文件
下列文件对于本文件的应用是必不可少的。凡是注日期的引用文件,仅注日期的版本适用于本文件。凡是不注日期的引用文件,其最新版本(包括所有的修改单)适用于本文件。
GB/T 8170 数值修约规则与极限数值的表示和判定
GB/T 12806实验室玻璃仪器 单标线容量瓶
GB/T 12808实验室玻璃仪器 单标线吸量管
GB/T 17617耐火原料抽样检验规则
3 仪器和设备
3.1 天平
感量 0.1mg。
3.2 恒温干燥箱
3.3铂坩埚或瓷坩埚
30mL.
3.4 分光光度计
3.5高温炉
最高使用温度≥1100℃,且能自动控温的箱式电炉。
3.6容量瓶
GB/T 12806 A类。
3.7 吸量管
GB/T 12808 A类。
3.8 气体容量法测量装置
3.8.1 锥形瓶
250mL,可从瓶口放入称量瓶。
3.8.2 称量瓶
容积约 10mL。
3.8.3 冷凝管
球形冷凝管,长度不小于 400mm。
3.8.4 三通活塞
异 T型毛细管三通活塞,活塞部分涂以真空油脂。
3.8.5 量气管
精度不低于 0.2mL。
3.8.6 水冷套
长约 460mm
3.8.7 参比管
内径5mm~10mm。
3.8.8 贮气瓶
700mL~800mL.
3.8.9 水准瓶
750mL~1000ml.
3.8.10 水浴槽深度不小于 150mm,可将 250ml,容量瓶浸没,带有出水口和入水口。
3.8.11 电炉盘
功率可调。
3.9 温度计
精度 0.5℃.
3.10 恒温水箱
在试验过程中水温变化不大于 1℃。
3.11 气压表
精度 0.01kPa。
4 试样制备
4.1 采样
按 GB/T 17617 采集实验室样品。
4.2 制备
4.2.1 将实验室样品破碎至 6.7mm 以下,按四分法缩分至约 100g.
4.2.2 当合同另有取样约定或由于产品形式的限制,无法取得>100g的实验室样品时,可以例外。
4.2.3将缩分后的样品粉碎至 0.5mm 以下,继续缩分,并加工成粒度小于 0.088mm 的试样。
4.2.4 试样分析前应在恒温干燥箱(3.2)中 105℃~110℃烘干至少 2h,并置于干燥器中冷至室温
5 通则
5.1 测定次数
在重复性条件下测定2次。
5.2 空自试验
在重复性条件下做空白试验,所用试剂必须取自同一试剂瓶。
5.3 结果表述
结果应按 GB/T 8170 修约,保留2位小数;如果委托方供货合同或有关标准另有要求时,可按要求的位数修约。
5.4 分析结果的采用
当2次有效分析值之差不大于表2、表3、表5所规定的允许差时,以其算术平均值作为最终分析结果;否则,应按附录 A的规定进行追加分析和数据处理。
5.5 质量保证和控制
5.5.1 标准曲线应定期(不超过3个月)用标准物质校准一次。如果仪器维修或者更换部件,应重新绘制工作曲线,并用同类型标准物质校准。当标准物质的分析值与标准值之差大于表 2、表 3、表5所规定允许差的 0.7倍时,应重新绘制标准曲线。
5.5.2 仲裁试验时,应随同试样分析同类型标准物质。当标准物质的分析值与标准值之差不大于表 2、表 3、表5 所规定允许差的 0.7倍时,则试样分析值有效,否则无效。
6 试验报告
试验报告应至少包括以下内容:
-委托单位;
-试样名称;
-分析结果;
-使用标准;
-与规定的分析步骤的差异(如有必要);
-在试验中观察到的异常现象(如有必要);
-试验日期。
7单质硅的测定
7.1 稀碱分离法[20%<w(Si)<97%]
7.1.1 原理本方法适用于不含可溶于氢氧化钠溶液的硅化合物的工业硅制品。
试样用氢氧化钠溶液浸煮,其中的单质硅转换成为可溶性的硅酸盐,二氧化硅不溶解。分离碱不溶物后,加盐酸酸化滤液并完全蒸干。用聚氧化乙烯凝聚硅酸,过滤并灼烧成二氧化硅。然后用氢氟酸处理灼烧后的残渣。氢氟酸处理前后的质量差即为二氧化硅主量。再用熔剂处理挥硅后的残渣,稀盐酸浸取,并入滤液。用钼蓝光度法测定溶液中的二氧化硅量。两者相加之和即为单质硅转换成的二氧化硅总量。最后将其换算成单质硅含量。
7.1.2 试剂
7.1.2.1氢氧化钠溶液(50g/L):贮存于塑料瓶中。
7.1.2.2聚氧化乙烯溶液(1.0g/L):取 0.2g聚氧化乙烯溶解在 200mL水中。有效期两周。
7.1.2.3盐酸(p=1.19g/mL)。
7.1.2.4 盐酸(1+1)。
7.1.2.5盐酸(5+95)。
7.1.2.6硝酸银溶液(10g/L)。
7.1.2.7氢氟酸(p=1.14g/mL)。
7.1.2.8钼酸铵溶液(50g/L):过滤后使用。
7.1.2.9草酸一硫酸混合酸:称取 25g 草酸,溶于 250ml,硫酸(1+8)溶解,用水稀释至 1000mL,混匀。
7.1.2.10 硫酸亚铁铵溶液:称取 50g 硫酸亚铁铵溶解在一定量水中,加 10mL 硫酸(1+1),最后用水稀
至 1000mL,混匀,过滤后使用。用时配制。
7.1.2.11 硫酸(p=1.42g/mL)。
7.1.2.12 对硝基苯酚(5g/L):用乙醇配制。
7.1.2.13 氢氧化钠溶液(200g/L):贮存于塑料瓶中,
7.1. 2. 14盐酸(1+4)。
7.1.2.15 混合熔剂:两份无水碳酸钠与一份硼酸研细,混匀。
7.1.2.16 二氧化硅标准贮存溶液(含 SiO,0.5mg/ml):称取 0.1000g预先在 1000℃灼烧 2h 并冷却至室温的二氧化硅(纯度 99.99%)于铂坩埚中,加人2g~3g无水碳酸钠(基准试剂),盖上坩埚盖并稍留缝隙,置于 1000℃高温炉中熔融 5min~10min,取出冷却。置于盛有 100ml,沸水的聚四氟乙烯烧杯中,低温加热浸取熔块至试液清亮,用热水洗出坩埚及盖,冷却至室温。移人 200mL,容量瓶中,用水稀释至刻度,摇匀,贮存于干燥过的洁净塑料瓶中。
7.1.2.17 二氧化硅标准贮存溶液(含 SiO:50μg/mL):移取 25.00ml,二氧化硅贮存液(7.1.2.16)到 250mL,容量瓶中,用水稀释至刻度,摇匀,再转移至干燥过的洁净的塑料瓶中。
7.1.2.18 二氧化硅标准贮存溶液(含 SiO,5μg/mL):
移取 25.00ml,二氧化硅贮存液(7.1.2.17)到 250ml,容量瓶中,用水稀释至刻度,摇匀,再转移至干燥过的洁净的塑料瓶中。
7.1.3 试样量
称取约 0.2g 试样,精确至 0.1mg.
7.1.4 测定
7.1.4.1 称取试样于朔料烧杯中,加人 40mL,氢氧化钠溶液(7.1.2.1),盖上表面皿,于水浴锅上浸者(水浴锅开始温度不宜过高,一般不高于 50℃,浸煮温度为 100℃),待试样几乎反应完全后,用小片定量滤纸仔细擦洗表面皿和烧杯内壁,并用热水冲洗,再继续浸煮 1h(可以不盖表面皿),每 15min 搅拌一次,取下冷却。用中速定量滤纸过滤,滤液承接于 400mL,烧杯中。以近沸腾的热水冲洗滤纸及残渣至中性。残渣及滤纸保留,放入铂坩埚中低温灰化(以测定试样中的二氧化硅用)。
7.1.4.2 滤液加盐酸(7.1.2.3)10ml~15ml,于电热板上完全蒸干,取下冷却后加 10ml 盐酸(7.1.2.3)、10mL,聚氧化乙烯溶液(7.1.2.2),用 10ml,热水冲洗烧杯内壁及玻璃棒,充分搅拌 1min~2min 后,再次用 10ml,热水冲洗烧杯内壁及玻璃棒,在 50℃~70℃电热板上保温 10min~15min,使可溶性盐类完全溶解。以慢速定量滤纸过滤。滤液承接于 250mL, 容量瓶 A 中。用热盐酸(7.1.2.5)将烧杯中的沉淀转移到漏斗上。用小片定量滤纸仔细擦洗玻璃棒及烧杯内壁,并一同转移到漏斗中。再用热水洗涤沉淀至无氯离子为止(用硝酸银溶液(7.1.2.6)检查)。
7.1.4.3 沉淀连同滤纸(7.1.4.2)置于铂坩埚中,小心烘干、灰化,放人高温炉中,从低温开始,逐渐升温至 1000℃~1050℃左右并灼烧 1h,将其取出,置于干燥器中冷却至室温,称量。如此反复操作(每次灼烧15min)直至恒重(m)。
7.1.4.4 灼烧后的沉淀,用水润湿,加1滴~2 滴硫酸(7.1.2.11)、5mL,氢氟酸(7.1.2.7),低温蒸发至冒硫酸白烟,取下稍冷,再加 2mL~3mL,氢氟酸(7.1.2.7),继续蒸发至干,此时稍升高温度,小心加热除尽硫酸白烟,然后置于高温炉内 1000℃~1050℃灼烧 30min,将其取出,置于干燥器中,冷却至室温,称量。如此反复操作(每次灼烧 15min),直至恒重(m2)。
7.1.4.5 加约 1g的混合熔剂(7.1.2.15)到烧后的铂坩埚(7.1.4.4)中,置于 1000℃~1050℃的高温炉中熔融 5min。取出冷却加 5mL, 盐酸(7.1.2.4)浸取,合并到承接原滤液的容量瓶 A中,用水稀释至刻度,摇匀。
7.1.4.6 从容量瓶 A 中分取 10.00mL,试液于 100mL,塑料烧杯中,加入1滴对硝基苯酚(7.1.2.12),用氢氧化钠(7.1.2.13)调至黄色,再加人 2mL,盐酸(7.1.2.14)。将溶液移人 100mL, 容量瓶中
7.1.4.7 加人 5ml,钼酸铵(7.1.2.8),摇匀,于室温下放置 20min~30min(室温低于 15℃则在约 30℃的温水中进行)。加人 25ml,草酸一硫酸混合液(7.1.2.9),摇匀,放置 2min。加人5mL,硫酸亚铁铵溶液(7.1.2.10),用水稀释至刻度,摇匀。
7.1.4.8 用 30mm 吸收皿,于分光光度计波长 810nm 处,以空白试验溶液为参比测量其吸光度。
7.1.5 工作曲线的绘制用滴定管移取 0mL、1.00mL、2.00ml、4.00ml、6.00mL、8.00mL、10.00mL 二氧化硅标准溶液(7.1.2.18),分别于一组 100ml,容量瓶中,加人 2mL盐酸(7.1.2.14),加水至约 20mL~30mL。之后按照 7.1.4.7和 7.1.4.8操作。以试剂空白为参比测其吸光度,绘制工作曲线。
7.1.6 分析结果的计算
单质硅量用质量分数 u(Si)计,数值以%表示,按式(1)计算:
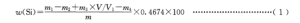
式中:
m–试料质量的数值,单位为克(g);
m1–氢氟酸处理前沉淀与坩埚的质量的数值,单位为克(g);
m2–氢氟酸处理后沉淀与坩埚的质量的数值,单位为克(g);
m3–由工作曲线査得的二氧化硅量的数值,单位为克(g);
m4–重量法空白试验的二氧化硅的质量的数值,单位为克(g);
V1–分取试液的体积的数值,单位为毫升(mL);
V-试液总体积的数值,单位为毫升(mL);
0.4674–二氧化硅与单质硅换算系数。
7.1.7 允许差
该方法两次有效分析值之差应不大于表2规定的允许差。
|
表2稀碱分离法单质硅分析值允许差 |
|
|
w(Si)/% |
允许差/% |
|
>20~≤50 |
0.30 |
|
>50~≤80 |
0.50 |
|
>80~≤97 |
0.60 |
7.2 气体容量法[0.5%≤w(Si)<99%]
7.2.1原理
试料在氢氧化钠溶液中煮沸,其中的单质硅按 ![]() 反应产生氢气氢气近似于理想气体。测量产生氢气的体积、温度和压强,根据理想气体状态方程及反应方程式,计算出单质硅的含量。
反应产生氢气氢气近似于理想气体。测量产生氢气的体积、温度和压强,根据理想气体状态方程及反应方程式,计算出单质硅的含量。
7.2.2 试剂
7.2.2.1 氢氧化钠溶液(200g/L)。
7.2.2.2 盐酸(1+1)。
7.2.2.3 封闭液:在 1000m 硫酸(1+1000)中滴加几滴甲基橙(1.0g/L)至淡红色
7.2.3 测量装置
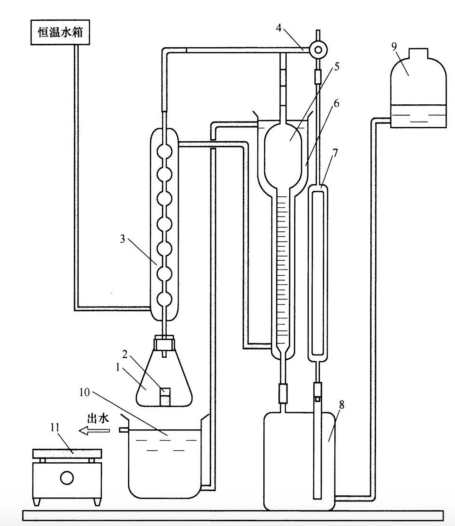
气体容量法测量装置示意图见图 1。贮气瓶和水准瓶内装人适量封闭液。整个装置在试验过程中不允许漏气和漏水。
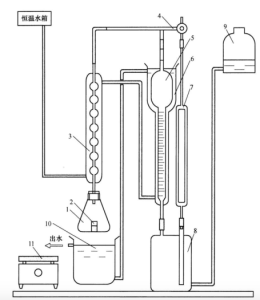
说明:
1-锥形瓶;
2-称量瓶;
3-冷凝管;
4-三通活塞;
5-量气管;
6-水冷套;
7-参比管;
8-贮气瓶;
9-水准瓶;
10-水浴槽;
11-电炉盘
图1 气体容量法测量装置示意图
试样量
根据单质硅的大致含量,按公式(2)计算称样量,控制最小称样量不低于 0.1g,最大称样量不超过6g,称样精确至 0.1mg.
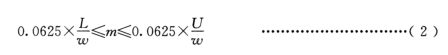
式中:
m-称样量,单位为克(g);-
w-单质硅质量百分数,单位为百分数(%);
L-量气管低刻度值,单位为毫升(mL);
U –量气管高刻度值,单位为亳升(mL);
0.0625–标准状态下单位质量单质硅产生的氢气体积(g/mL)。
7.2.5 测定
7.2.5.1 测量装置的准备和检查将空锥形瓶连接在图1所示的测量装置上。将三通活塞置于三通位置,调节水准瓶位置,使封闭液液面位于量气管零刻线处。顺时针旋转三通活塞 135°,使之位于三向密封位置,将水准瓶降至最低位置保持 2min~3min,观察量气管,参比管中封闭液的液面变化,若参比管内液面位于零刻线水平位置不变量气管内液面高于水准瓶液面且稳定不变,可以确认测量装置的气密性符合要求。
7.2.5.2 试样中金属铝等干扰的消除用称量瓶直接称取试样。缓慢往称量瓶中加人 5mL,盐酸(7.2.2.2),轻轻摇动,使试料与盐酸充分接触。将称量瓶置于低温电热板(≤130℃)上加热,观察是否持续有均匀小气泡冒出,倘若在蒸干前持续有均匀小气泡冒出,再加入 5mL,盐酸(7.2.2.2),继续加热。重复上述操作,直至无小气泡冒出。取下称量瓶,冷却至室温,向其注人去离子水至称量瓶刻线处,
7.2.5.3 装样向锥形瓶中加人 100ml,氢氧化钠溶液(7.2.2.1)。用镊子将按 7.2.5.2处理后的盛有试样的称量瓶置于锥形瓶中,保证称量瓶中的试料不与锥形瓶中的氢氧化钠溶液接触,按图1所示将锥形瓶连接到测量装置上,并保证连接牢固、不气。
7.2.5.4 初始态参数的确定
7.2.5.4.1 将锥形瓶浸人水浴槽中。开启恒温水流冷却装置。待水浴槽中的水温与水冷套中的水温一致后,再保持 5min。
7.2.5.4.2 将三通活塞置于三通位置,调节水准瓶高度,使量气管的液面稳定于零刻线处。然后固定水准瓶位置,将三通活塞顺时针转动 135°,使反应装置处于气密状态。
7.2.5.4.3 读取水浴槽中或水冷套中温度计显示的温度,换算成开尔文温度,以此作为量气管内气体初始状态的温度 T。
7.2.5.5 加热反应
7.2.5.5.1 撤去水浴槽,擦干锥形瓶外壁。将锥形瓶连同冷凝管一起倾斜,使称量瓶倾倒,试样浸人至锥形瓶内的氢氧化钠溶液中。
7.2.5.5.2 将电炉盘置于锥形瓶下方通电加热。待锥形瓶中的溶液沸腾后,调低电炉功率,保持微沸状态。在此过程中,因有大量氢气的产生,量气管中封闭液的液面会急剧降低,应注意及时降低水准瓶的高度,使水准瓶与量气管(或贮气瓶)的液面高差保持在 50mm 以内。7.2.5.5.3 待量气管(或贮气瓶)内的封闭液的液面基本不再变化时,再持续加热 5min~10min,然后停止加热,撤去电炉盘。一般总加热时间大约在 30min~80min 之间,即可反应完全。7.2.5.6 终态参数和表观气体产生量的确定
7.2.5.6.1 将加热反应后的锥形瓶再次浸入水浴槽中进行冷却。在冷却过程中要及时调整水准瓶的高度,使量气管内的液面与水准瓶的液面基本保持水平。
7.2.5.6.2 待水浴槽内水温与水冷套内水温一致时,固定水准瓶的位置,继续冷却约 5min~10min。若最后 5min 内,量气管的液面变化不超过 0.2ml,时,将三通活塞顺时针转动 45,使参比管与大气连通。
7.2.5.6.3 调节水准瓶的高度,使量气管,参比管内的液面位于同一水平线上,固定水准瓶,从量气管液面处读取表观气体产生量V,精确至 0.2mL。
7.2.5.6.4 读取水冷套内温度计显示的温度,换算成开尔文温度,以此作为量气管内气体的终态温度 T。
7.2.5.6.5 用气压表测量大气压值 P精确至 0.01kPa。应注意按仪器使用说明书进行修正。
7.2.5.6.6 将三通活塞旋至三通位置,关闭恒温水流冷却装置,取下锥形瓶,结束试验。
7.2.5.7 真实气体产生量的确定
当始态和终态冷却水温变化|T0-T1|≤1.0K 时,以表观气体产生量V1 作为真实气体产生量 V0。若试验设施无法达到|T0-T1|≤1.0K条件时,真实气体产生量V按公式(3)计算:
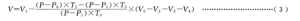
式中:
V0-表观气体产生量,单位为毫升(mL);
Vi-实验室内大气压,单位为千帕(kPa);
T0,T1-始、终态水浴槽或水冷套内的水温,单位为开尔文(K);
P0,P1,–温度 T。、T,时纯水的饱和蒸汽压,单位为千帕(kPa);
V0–测量装置内锥形瓶至量气管零刻度线间的总有效体积,单位为毫升(mL);
V2锥形瓶内加入的氢氧化钠溶液(7.2.2.1)的体积,单位为毫升(mL);
V3称样器中试样、盐酸及去离子水的体积,单位为毫升(mL);
V4柱形称样器浸人氢氧化钠溶液部分的体积,单位为毫升(mL)。
7.2.6 结果计算
单质硅量用质量分数 w(Si)计,数值以%表示,按式(4)计算:
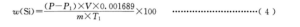
式中
P-实验室内的大气压,单位为千帕(kPa);
P1-温度为T1时纯水的饱和蒸汽压,单位为千帕(kPa);
V-真实气体产生量,单位为毫升(mL);
m称取的试样量,单位为克(g);
T1-终态下量气管内气体的温度,单位为开尔文(K);
0.001689–氢气体积换算成单质硅质量的系数。
7.2.7 允许差
该方法两次有效分析值之差应不大于表3规定的允许差。
|
表3气体容量法单质硅分析值允许差 |
|
|
w(Si)/% |
允许差/% |
|
>0.5~≤5 |
0.10 |
|
>5~≤10 |
0.20 |
|
>10~≤99 |
0.50 |
8 二氧化硅的测定
8.1 总则
本方法不适用于含有碳化硅的工业硅制品,
二氧化硅的测定可以按照以下两种方法进行:
a) 钼蓝光度法(0.5%~10%)(8.2);
b)重量-钼蓝光度法(>10%)(8.3)。
8.2 钼蓝光度法(0.5%~10%)
8.2.1 原理
试样经氢氧化钠溶液浸煮过后,二氧化硅不溶解。分离碱不溶物后,将残渣用混合熔剂熔融,用钼蓝光度法测定溶液中的二氧化硅量。
8.2.2 试剂
8.2.2.1 混合熔剂:两份无水碳酸钠与一份硼酸研细,混匀。
8.2.2.2 钼酸铵溶液(50g/L):过滤后使用。
8.2.2.3草酸一硫酸混合酸:称取 25g 草酸,溶于 250ml 硫酸(1+8)溶解,用水稀释至 1000mL,混匀。
8.2.2.4 硫酸亚铁铵溶液(50g/L):称取 50g硫酸亚铁铵溶解在一定量水中,加 10mL 硫酸(1+1),最后用水稀释至 1000mL,混匀,过滤后使用。用时配制。
8.2.2.5 盐酸(1+4)。
8.2. 2.6无水乙醇。
8.2.2.7 二氧化硅标准贮存溶液(含 SiO,0.5mg/ml):称取 0.1000g预先在 1000℃灼烧 2h 并冷却至室温的二氧化硅(纯度 99.99%)于铂坩埚中,加人 2g~3g无水碳酸钠(基准试剂),盖上坩埚盖并稍留缝隙,置于 1000℃高温炉中熔融 5min~10min,取出冷却。置于盛有 100mL,沸水的聚四氟乙烯烧杯中,低温加热浸取熔块至试液清亮,用热水洗出坩埚及盖,冷却至室温。移人 200mL,容量瓶中,用水稀释至刻度,摇匀,贮存于干燥过的洁净塑料瓶中。
8.2.2.8 二氧化硅标准贮存溶液(含 Si0,50μg/mL):移取 25.00ml 二氧化硅贮存液(8.2.2.7)移人250mL, 容量瓶中,用水稀释至刻度,摇匀,贮存于干燥过的洁净塑料瓶中。
8.2.3 试样量
称取约 0.2g试样,精确至 0.1mg。
8.2.4 测定
8.2.4,1 将经过氢氧化钠溶液(7.1.2.1)浸煮并过滤后的滤纸及残渣(7.1.4.1),放入铂坩埚中,烘干、灰化,加人混合熔剂 1g~2g,放人 800℃~900℃高温炉中,升温至 1000℃~1050℃熔融 5min~15min,取出,冷却。
8.2.4.2 在 200mL,烧杯中加入热的 100mL 盐酸(8.1.2.5)将熔融物低温浸取出来,冷却,移入 200ml容量瓶,用水稀释至刻度。摇匀。
8.2.4.3 移取 10.00mL, 试液(8.2.4.2)[试样中 w(Si0,)>5%时,则移取 5.00mL,试液,加 5mL,空白试
液]于 100mL 容量瓶中。8.2.4.4 加人 10mL,无水乙醇(8.2.2.6)、5ml 钼酸铵溶液(8.2.2.2),摇匀,于室温下放置 20min~30min(室温低于 15℃则在约 30℃的温水浴中进行)。8.2.4.5 加人 25m, 草酸-硫酸混合溶液(8.2.2.3),摇匀,放置 2min,加人 5ml 硫酸亚铁铵溶液(8.2.2.4),用水稀释至刻度,摇匀。
8.2.4.6 用合适的吸收皿(见表4),于分光光度计波长 690nm 处,以空白试验溶液为参比测量其吸光度。
|
表4按二氧化硅的含量选择吸收皿 |
||
|
w(SiO₂)/% |
0.5~2.5 |
2.5~10 |
|
吸收皿/mm |
10 |
5 |
|
标准曲线 |
8.2.5 |
8.2.5 |
8.2.5 标准曲线的绘制移取 0mL、1.00mL、2.00mL、4.00mL、6.00mL、8.00mL、10.00ml 二氧化硅标准溶液(8.2.2.8)分别置于一组 100ml 容量瓶中,加 2mL, 盐酸(8.2.2.5),加水至 20ml。以下按 8.2.4.4~8.2.4.6 操作。以试剂空白为参比测量其吸光度,绘制标准曲线。
8.2.6 分析结果的计算
二氧化硅量用质量分数 w(SiO,)计,数值以%表示,按式(5)计算:
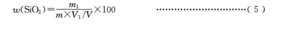
式中:
m1-由标准曲线査得的二氧化硅量的数值,单位为克(g);
m-试料质量的数值,单位为克(g);
V1-分取试液的体积的数值,单位为毫升(mL);
V-试液总体积的数值,单位为毫升(mL)。
8.3 重量-钼蓝光度法(>10%)
8.3.1 原理
试样经氢氧化钠溶液浸煮过后,二氧化硅不溶解。分离碱不溶物后,将残渣用混合熔剂熔融,完全蒸干后用聚氧化乙烯凝聚析出硅酸,过滤并灼烧成二氧化硅。用氢氟酸处理灼烧物。氢氟酸处理前后的质量差即为二氧化硅主量。再用熔剂熔融氢氟酸处理后的残渣,稀盐酸浸取,并人滤液,用钼蓝光度法测定溶液中的残余二氧化硅量。两者相加即为试样中二氧化硅的量。8.3.2 试剂
8.3.2.1 混合熔剂:两份无水碳酸钠与一份硼酸研细,混匀。
8.3.2.2 钼酸铵溶液(50g/1):过滤后使用。8.3.2.3草酸一硫酸混合酸:称取 25g 草酸,用 250mL 硫酸(1+8)溶解,用水稀释至 1000mL,混匀。8.3.2.4 硫酸亚铁铵溶液(50g/L):称取 50g硫酸亚铁铵溶解在一定量水中,加 10mL 硫酸(1+1),最后用水稀释至 1000mL,混匀,过滤后使用。用时配制。
8.3.2.5 对硝基苯酚(5g/L):用乙醇配制。
8.3.2.6 氢氧化钠溶液(200g/L):贮存于塑料瓶中。
8.3. 2.7盐酸(p=1.19g/mL)。
8.3. 2.8硫酸(p=1.42g/mL)
8.3.2.9 氢氟酸(p=1.13g/mL)。
8.3.2.10 盐酸(1+4)
8.3. 2. 11盐酸(1+1),
8.3.2.12 盐酸(5+95)
聚氧化乙烯(1g/1):称取 0.2g聚氧化乙烯溶解在 200m,水中。
8.3. 2. 13有效期两周。
8.3. 2. 14硝酸银:10g/L。
8.3.2.15 二氧化硅标准贮存溶液(含 SiO,0.5mg/mL):称取 0.1000g预先在 1000℃灼烧 2h 并冷却至室温的二氧化硅(纯度 99.99%)于铂坩埚中,加人 2g~3g无水碳酸钠(基准试剂),盖上坩埚盖并稍留缝隙,置于 1000℃高温炉中熔融 5min~10min,取出冷却。置于盛有 100ml,沸水的聚四氟乙烯烧杯中,低温加热浸取熔块至试液清亮,用热水洗出坩埚及盖,冷却至室温。移人 200mL,容量瓶中,用水稀释至刻度,摇匀,贮存于干燥过的洁净塑料瓶中。
8.3.2.16 二氧化硅标准贮存溶液(含SiO,50μg/ml):移取 25.00ml,二氧化硅贮存液(8.3.2.15)移入250mL, 容量瓶中,用水稀释至刻度,摇匀,贮存于干燥过的洁净塑料瓶中。
8.3.2.17 二氧化硅标准贮存溶液(含 SiO,5g/mL):移取 25.00m 二氧化硅贮存液(8.3.2.16)移人250ml, 容量瓶中,用水稀释至刻度,摇匀,贮存于干燥过的洁净塑料瓶中。
8.3.3 试样量
称取约 0.2g 试样,精确至 0.1mg。
8.3.4.1 将经过氢氧化钠溶液(7.1.2.1)浸煮并过滤后的滤纸及残渣(7.1.4.1),放入铂坩埚中,烘干灰化,加人混合熔剂 1g~2g,放人 800℃~900℃高温炉中,升温至 1000℃~1050℃熔融 5min~15min,取出,冷却。
8.3.4.2 在 200ml,烧杯中放入煮沸的 40ml,盐酸(8.3.2.11)和 50mL 水。将熔融物加热浸取出来。低温加热并完全蒸干,取下冷却后加 10ml 盐酸(8.3.2.7)、10mL,聚氧化乙烯(8.3.2.13),用 10ml 热水冲洗烧杯内壁及玻璃棒,充分搅拌 1min~2min后,再次用 10ml,热水冲洗烧杯内壁及玻璃棒,在 50℃~70℃电热板上保温 10min~15min,使可溶性盐类完全溶解。以慢速定量滤纸过滤。滤液承接于 250ml容量瓶 B中。用热盐酸(8.3.2.12)将烧杯中的沉淀转移到漏斗上。用小片滤纸擦洗烧杯内壁及玻璃棒,一并转移到漏斗中。再用热水洗涤至无氯离子为止(用硝酸银溶液(8.3.2.14)检查)。
8.3.4.3 沉淀连同滤纸(8.3.4.2)置于铂坩埚中,烘干、灰化。放入 1000℃~1050℃高温炉灼烧 1h,将其取出,置于干燥器中冷却至室温,称量。如此反复操作(每次灼烧 15min)直至恒重(m)。8.3.4.4 灼烧后的沉淀,用水润湿,加1滴~2 滴硫酸(8.3.2.8)、5m,氢氟酸(8.3.2.9),低温蒸发至冒硫酸白烟,取下稍冷,再加 2ml,~3mL, 氢氟酸(8.3.2.9),继续蒸发至干,此时稍升高温度,小心加热除尽硫酸白烟,然后置于高温炉内,1000℃~1050℃左右灼烧 30min,将其取出,置于干燥器中,冷却至室温,称量。如此反复操作(每次灼烧 15min),直至恒重(m)。
8.3.4.5 加约 1g的混合熔剂(8.3.2.1)到烧后的铂金坩埚中(8.3.4.4),置于 1000℃~1050℃的高温炉中熔融 5min~15min。取出冷却加 5ml,盐酸(8.3.2.11)浸取,合并到承接原滤液的容量瓶B中,用水稀释至刻度,摇匀。
8.3.4.6 从容量瓶 B 中分取 10.00mL, 试液于 100mL,塑料烧杯中,加入1滴对硝基苯酚(8.3.2.5),用氢氧化钠(8. 3.2.6)调至黄色,再加人 2mL 盐酸(8.3.2.10)。将溶液移人 100ml, 容量瓶中。
8.3.4.7 加人 5ml,钼酸铵(8.3.2.2),摇匀,于室温下放置 20min~30min(室温低于 15℃则在约 30℃的温水中进行)。加入 25ml,草酸-硫酸混合液(8.3.2.3),摇匀,放置 2min。加人 5ml,硫酸亚铁铵溶液(8.3.2.4),用水稀释至刻度,摇匀。
8.3.4.8 用30mm 吸收皿,于分光光度计波长 810nm 处,以空白试验溶液为参比测量其吸光度。
8.3.5 工作曲线的绘制用滴定管移取 0mL、1.00mL、2.00mL、4.00mL、6.00mL、8.00ml、10.00mL 二氧化硅标准溶液(8.3.2.17),分别于一组 100ml, 容量瓶中,加入2ml盐酸(8.3.2.10),加水至约 20mL~30mL。以下按照 8.3.4.7 和 8.3.4.8操作。以试剂空白为参比测其吸光度,绘制工作曲线。
8.3.6 分析结果的计算
二氧化硅量用质量分数 w(SiO,)计,数值以%表示,按式(6)计算:
![]()
式中:
m-试料质量的数值,单位为克(g);
m1-氢氟酸处理前沉淀与坩埚的质量的数值,单位为克(g);
m2-氢氟酸处理后沉淀与坩埚的质量的数值,单位为克(g);
m3-由工作曲线查得的二氧化硅量的数值,单位为克(g);
m4-重量法空白试验的二氧化硅的质量的数值,单位为克(g);V–分取试液的体积的数值,单位为毫升(ml);
V-试液总体积的数值,单位为亳升(mL)。
8.4 允许差
该方法两次有效分析值之差应不大于表5规定的允许差。
Chemical analysis of elemental silicon and silicon dioxidein industrial silicon for refractories
1 Range
This standard specifies methods for chemical analysis of elemental silicon and silica in industrial silicon for refractories.
This standard is applicable to the determination of elemental silicon and silicon dioxide content in industrial silicon for refractory materials, the determination range is shown in Table 1.
The determination of elemental silicon content in refractories can also refer to this standard.
|
Table 1 Measurement range |
|
|
Analysis item |
Range (mass score)/% |
|
Si |
20~97(Dilute alkali separation method) |
|
0.5~99(gas volume method) |
|
|
SiO2 |
>0.5 |
2 Normative reference documents
The following documents are essential for the application of this document. For dated references, the date-only version applies to this document. For undated references, the latest version (including all amendment orders) applies to this document.
GB/T 8170 numerical revision rules and limit value representation and judgment
GB/T 12806 Laboratory glass instrument single-line volumetric bottle
GB/T 12808 Laboratory glass instrument single-line pipette
GB/T 17617 Rules for sampling inspection of refractory raw materials
3 Instruments and equipment
3.1 Balance
Sensitivity is 0.1mg.
3.2 Constant temperature drying oven
3.3 Platinum or porcelain crucible
30mL.
3.4 Spectrophotometer
3.5 High temperature furnace
Box type electric furnace with maximum operating temperature ≥1100℃ and automatic temperature control.
3.6 Volumetric bottle
GB/T 12806 Class A.
3.7 Pipette
GB/T 12808 Class A.
3.8 Gas capacity method measuring device
3.8.1 Conical bottle
250mL, can be put into the weighing bottle from the mouth of the bottle.
3.8.2 Weighing bottle
The volume is about 10mL.
3.8.3 Condensate Pipe
Spherical condensing tube, length not less than 400mm.
3.8.4 Tee Piston
Different T type capillary tee piston, piston part coated with vacuum grease.
3.8.5 Measure the trachea
Accuracy is not less than 0.2mL.
3.8.6 Water-cooled jacket
The length is about 460mm
3.8.7 Reference tube
Inner diameter 5mm~10mm.
3.8.8 Gas cylinder
700mL~800mL.
3.8.9 Level bottle
750mL~1000ml.
3.8.10 The depth of the water bath is not less than 150mm, and the 250ml volumeter bottle can be submerged with water outlet and water inlet.
3.8.11 Electric Stove Plate
The power is adjustable.
3.9 Thermometer
Accuracy 0.5℃.
3.10 Thermostatic water tank
During the test, the change of water temperature is not more than 1℃.
3.11 Barometer
Accuracy: 0.01kPa.
4 Sample preparation
4.1 Sampling
Laboratory samples were collected at GB/T 17617.
4.2 Preparation
4.2.1 The laboratory sample shall be broken to less than 6.7mm and divided into about 100g according to the quarter method.
4.2.2 Exceptions may be made when the contract provides otherwise for sampling or when it is not possible to obtain laboratory samples of >100g due to limitations in the form of the product.
4.2.3 The sample after reduction is crushed to less than 0.5mm, continued reduction, and processed into a sample with a particle size of less than 0.088mm.
4.2.4 Before analysis, the sample should be dried at 105℃~110℃ in a constant temperature drying oven (3.2) for at least 2h, and placed in a dryer to cool to room temperature
5 General Rules
5.1 Measurement times
It was measured twice under the condition of repeatability.
5.2 Air self test
To perform blank tests under repeatable conditions, reagents used must be taken from the same reagent bottle.
5.3 Result Presentation
The result should be modified according to GB/T 8170, with 2 decimal places reserved; If the principal’s supply contract or relevant standards require otherwise, the contract can be modified according to the required digit.
5.4 Adoption of analysis results
When the difference between the two effective analysis values is not greater than the allowable difference specified in Table 2, Table 3 and Table 5, the arithmetic average of the two effective analysis values shall be taken as the final analysis result. Otherwise, additional analysis and data processing shall be carried out in accordance with Appendix A.
5.5 Quality assurance and control
5.5.1 The standard curve shall be calibrated periodically (not more than 3 months) with a standard material. If the instrument is repaired or parts are replaced, the working curve should be redrawn and calibrated with the same type of standard material. When the difference between the analytical value of the standard material and the standard value is greater than 0.7 times the allowable difference specified in Table 2, Table 3, Table 5, the standard curve should be redrawn.
5.5.2 When arbitrating the test, the same type of reference material shall be analyzed with the sample. When the difference between the analytical value of the standard material and the standard value is not greater than 0.7 times of the allowable difference stipulated in Table 2, Table 3 and Table 5, the analytical value of the sample is valid, otherwise it is invalid.
6 Test report
The test report shall include at least the following:
– the entrusted unit;
– Sample name;
– Analysis results;
– Use standards;
– Differences from the prescribed analysis steps (if necessary);
– Anomalies observed during the test (if necessary);
– Trial date.
7 Determination of elemental silicon
7.1 Dilute alkali separation [20%<w(Si)<97%]
7.1.1 Principle This method is applicable to industrial silicon products that do not contain silicon compounds soluble in sodium hydroxide solution.
The sample is soaked in sodium hydroxide solution, in which the elemental silicon is converted into soluble silicate, and the silica is not dissolved. After the alkali insoluble is separated, the filtrate is acidified with hydrochloric acid and completely dried. Silica is condensed with polyoxyethylene, filtered and burned to produce silica. The burned residue is then treated with hydrofluoric acid. The quality difference before and after hydrofluoric acid treatment is the main amount of silica. Then the residue after silicon removal is treated with a flux, diluted hydrochloric acid is leached and incorporated into the filtrate. The content of silica in solution was determined by molybdenum blu-ray method. The sum of the two is the total silicon dioxide converted from elemental silicon Amount. Finally, it is converted to elemental silicon content.
7.1.2 Reagents
7.1.2.1 Sodium hydroxide solution (50g/L): Store in plastic bottles.
7.1.2.2 Polyoxyethylene solution (1.0g/L): Take 0.2g polyoxyethylene and dissolve it in 200mL water. Valid for two weeks.
7.1.2.3 Hydrochloric acid (p=1.19g/mL).
7.1.2.4 Hydrochloric acid (1+1).
7.1.2.5 Hydrochloric acid (5+95).
7.1.2.6 Silver nitrate solution (10g/L).
7.1.2.7 Hydrofluoric acid (p=1.14g/mL).
7.1.2.8 Ammonium molybdate solution (50g/L): Use after filtration.
7.1.2.9 Oxalic acid and sulfuric acid mixture: Weigh 25g oxalic acid, dissolve in 250ml, sulfuric acid (1+8) dissolve, dilute with water to 1000mL, mix well.
7.1.2.10 Ammonium ferrous sulfate solution: Weigh 50g ammonium ferrous sulfate and dissolve it in a certain amount of water, add 10mL sulfuric acid (1+1), and finally dilute it to 1000mL with water, mix it well, and filter it before use. Use time to prepare.
Release to 1000mL, mix well, strain and use. Use time to prepare.
7.1.2.11 Sulfuric acid (p=1.42g/mL).
7.1.2.12 p-nitrophenol (5g/L): prepared with ethanol.
7.1.2.13 Sodium hydroxide solution (200g/L): stored in plastic bottles,
7.1. 2. 14 Hydrochloric acid (14 4).
7.1.2.15 Mixing flux: Grind two parts anhydrous sodium carbonate with one part boric acid and mix well.
7.1.2.16 Silica standard storage solution (including SiO,0.5mg/ml): Weigh 0.1000g silica that has been pre-calcined at 1000℃ for 2h and cooled to room temperature (purity 99.99%) in the platinum crucible, add 2g~3g anhydrous sodium carbonate (reference reagent), cover the crucible cover and leave a gap, melt in 1000℃ high temperature furnace for 5min~10min, remove and cool. Place in a teflon beaker containing 100ml boiling water, heat at low temperature to leach the frit until the test liquid is clear, wash the crucible and cover with hot water, and cool to room temperature. Transfer 200mL into a volumetric bottle, dilute to scale with water, shake well, and store in a dry clean plastic bottle.
7.1.2.17 Silica standard storage solution (including SiO:50μg/mL): Remove 25.00ml silica storage solution (7.1.2.16) to 250mL, dilute with water to the scale, shake well, and then transfer to a dry and clean plastic bottle.
7.1.2.18 Standard storage solution of silica (including SiO,5μg/mL):
Remove 25.00ml silica storage solution (7.1.2.17) to 250ml, dilute to scale with water, shake well, and transfer to dry clean plastic bottle.
7.1.3 Sample size
Weigh the sample about 0.2g, accurate to 0.1mg.
7.1.4 Determination
7.1.4.1 Weigh the sample in the plastic beaker, add 40mL, sodium hydroxide solution (7.1.2.1), cover the surface dish, and soak in the water bath (the starting temperature of the water bath should not be too high, generally not higher than 50℃, the immersion temperature is 100℃), after the sample is almost completely reacted, carefully scrub the surface dish and the inner wall of the beaker with a small piece of quantitative filter paper, rinse with hot water, continue to soak for 1h(can not cover the surface dish), stir every 15min, remove and cool. Filter with medium speed filter paper, filtrate in 400mL beaker. Rinse the filter paper and residue under nearly boiling hot water until neutral. The residue and filter paper are retained and put into a platinum crucible for low temperature ashing (to determine the silica in the sample).
7.1.4.2 Add 10ml to 15ml of hydrochloric acid (7.1.2.3) to the filtrate, completely dry on the electric heating plate, remove and cool, add 10ml of hydrochloric acid (7.1.2.3), 10mL of polyoxy-ethylene solution (7.1.2.2), rinse the inner wall of the beaker and glass rod with 10ml of hot water, and stir thoroughly After 1min to 2min, rinse the inner wall of the beaker and the glass rod with 10ml hot water again, and hold the beaker on the electric heating plate at 50℃ to 70℃ for 10min to 15min, so that the soluble salts are completely dissolved. Filter with slow filter paper. Filtrate is contained in 250mL volumetric bottle A. Transfer the precipitation from the beaker to the funnel with hot hydrochloric acid (7.1.2.5). Carefully scrub the glass rod and the inner wall of the beaker with a small piece of quantitative filter paper, and transfer them together to the funnel. Then wash with hot water and precipitate until no chloride ions are present (check with silver nitrate solution (7.1.2.6)).
7.1.4.3 Place the precipitation together with filter paper (7.1.4.2) in a platinum crucible, carefully dry and ash it, put it in a high temperature furnace, start from low temperature, gradually rise to about 1000℃~1050℃ and burn it for 1h, take it out, cool it in a dryer to room temperature, and weigh it. This operation is repeated (15min each time) until the constant weight (m).
7.1.4.4 After burning, moisten the precipitation with water, add 1 to 2 drops of sulfuric acid (7.1.2.11), 5mL, hydrofluoric acid (7.1.2.7), evaporate at low temperature until white sulfuric acid smoke, remove slightly cold, and then add 2mL~3mL, hydrofluoric acid (7.1.2.7), continue to evaporate until dry, at this time, slightly increase the temperature, carefully heat to remove the white sulfuric acid smoke, and then put it in a high temperature furnace 1000℃~1050℃ for 30min, remove it, put it in a dryer, cool to room temperature, weigh it. This operation is repeated (15min each time) until the constant weight (m2).
7.1.4.5 Add about 1g of mixed flux (7.1.2.15) to the burned platinum crucible (7.1.4.4), and melt for 5min in a high temperature furnace at 1000℃~1050℃. Remove and cool, add 5mL, leach with hydrochloric acid (7.1.2.4), combine into the volume bottle A to accept the original filtrate, dilute with water to the scale, shake well.
7.1.4.6 Divide 10.00mL from volumetric bottle A, add 1 drop of p-nitrophenol (7.1.2.12) into 100mL plastic beaker, adjust to yellow with sodium hydroxide (7.1.2.13), add 2mL, hydrochloric acid (7.1.2.14). Transfer the solution into a volume bottle of 100mL
7.1.4.7 Add 5ml of ammonium molybdate (7.1.2.8), shake well, and place at room temperature for 20min~30min(if the room temperature is lower than 15℃, do it in warm water at about 30℃). Add 25ml, oxalate-sulfuric acid mixture (7.1.2.9), shake well, set aside for 2min. Add 5mL, ammonium ferrous sulfate solution (7.1.2.10), dilute with water to scale, shake well.
7.1.4.8 The absorbance of a 30mm absorber was measured at the wavelength of 810nm by using a blank test solution as a reference.
7.1.5 For the drawing of the working curve, remove 0mL, 1.00mL, 2.00ml, 4.00ml, 6.00mL, 8.00mL, 10.00mL silica standard solution (7.1.2.18) from the burette and add it to a set of 100ml volumetric bottles 2mL hydrochloric acid (7.1.2.14), add water to about 20mL~30mL. Then follow 7.1.4.7 and 7.1.4.8. The absorbance was measured with reagent blank as reference, and the working curve was drawn.
7.1.6 Calculation of analysis results
Elemental silicon quantity is measured by mass fraction u(Si), the value is expressed as %, calculated according to formula (1) :
![]()
Formula:
m – the value of the mass of the sample, in grams (g);
m1– the value of the mass of precipitation and crucible before hydrofluoric acid treatment, in grams (g);
m2– The value of the mass of the precipitation and crucible after hydrofluoric acid treatment, in grams (g);
m3 The amount of silica measured by the working curve, in grams (g);
m4- The value of the mass of silica in the gravimetric blank test, in grams (g);
V1- the value of the volume of the test solution, in milliliters (mL);
V- The value of the total volume of the test liquid, in milliliters (mL);
0.4674– conversion coefficient between silica and elemental silicon.
7.1.7 Allowed difference
The difference between the two effective analysis values of the method should not be greater than the allowable difference specified in Table 2.
|
Table 2 Allowable difference in analytical value of elemental silicon by dilute alkali separation method |
|
|
w(Si)/% |
Allowable error/% |
|
>20~≤50 |
0.30 |
|
>50~≤80 |
0.50 |
|
>80~≤97 |
0.60 |
7.2 Gas capacity method [0.5%≤w(Si)<99%]
7.2.1 Principles
The sample is boiled in a solution of sodium hydroxide, in which the elemental silicon reacts with to produce hydrogen hydrogen, which is approximately an ideal gas. The volume, temperature and pressure of hydrogen produced are measured, and the content of elemental silicon is calculated according to the ideal gas state equation and reaction equation.
7.2.2 Reagents
7.2.2.1 Sodium hydroxide solution (200g/L).
7.2.2.2 Hydrochloric acid (1+1).
7.2.2.3 Sealing solution: Add a few drops of methyl orange (1.0g/L) to 1000m sulfuric acid (1+1000) until light red
7.2.3 Measuring Device
See Figure 1 for the schematic diagram of the gas volumetric measuring device. A proper amount of sealing liquid should be filled in the gas storage bottle and the leveling bottle. The whole device is not allowed to leak air and water during the test.
Instructions:
1- Conical bottle;
2- Weighing bottle;
3- Condensing tube;
4-tee piston;
5- Measuring trachea;
6- Water cooling sleeve;
7- Reference tube;
8- Gas cylinders;
9- Level bottle;
10- Water bath;
11- Electric stove plate
FIG. 1 Schematic diagram of gas volumetric measurement device
7.2.4 Sample size
According to the approximate content of elemental silicon, calculate the weighing sample amount according to formula (2), control the minimum weighing sample amount is not less than 0.1g, the maximum weighing sample amount is not more than 6g, the weighing sample is accurate to 0.1mg.
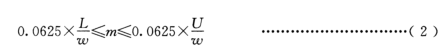
Formula:
m- sample size, expressed in grams (g); –
w- elemental silicon mass percentage, expressed in percentage (%);
L- Low scale value of the measuring pipe, in milliliters (mL);
U – measuring the high scale value of the gas pipe, the unit is mL;
0.0625- The volume of hydrogen produced per unit mass of elemental silicon under standard conditions (g/mL).
7.2.5 Measurement
7.2.5.1 Preparation and Inspection of the measuring device Connect the empty conical bottle to the measuring device shown in Figure 1. Put the tee piston in the tee position and adjust the position of the level bottle so that the sealing liquid level is located at the zero notch line of the measuring pipe. Turn the three-way piston 135° clockwise so that it is in the three-way sealing position and hold the level bottle to the lowest position 2min to 3min, observe the change of the liquid level of the sealing liquid in the measuring tube and the reference tube. If the liquid level in the reference tube is at the horizontal position of the zero marking line, and the constant liquid level in the trachea is higher than that of the level bottle, it can be confirmed that the air tightness of the measuring device meets the requirements.
7.2.5.2 Elimination of interference such as metal and aluminum in the sample directly weigh the sample with a weighing bottle. Slowly add 5mL of hydrochloric acid (7.2.2.2) into the weighing bottle and gently shake to make the test material fully in contact with hydrochloric acid. Place the weighing bottle on a low-temperature electric heating plate (≤130℃) to heat it and observe whether uniform small bubbles continue to emerge. If uniform small bubbles continue to emerge before drying, add 5mL and hydrochloric acid (7.2.2.2) and continue heating. Repeat until no small bubbles emerge. Remove the weighing bottle, cool to room temperature, fill it with deionized water to the marking of the weighing bottle,
7.2.5.3 Fill the conical bottle with 100ml sodium hydroxide solution (7.2.2.1). Place the weighing bottle containing the sample treated in accordance with 7.2.5.2 in a conical bottle with tweezers to ensure that the test material in the weighing bottle does not come into contact with the sodium hydroxide solution in the conical bottle. Connect the conical bottle to the measuring device as shown in Figure 1, and ensure that the connection is firm and non-air.
7.2.5.4 Determination of initial state parameters
7.2.5.4.1 Immerse the conical bottle in the human water bath. Turn on thermostatic water cooling. After the water temperature in the water bath is consistent with the water temperature in the water cooling jacket, keep it for 5 minutes.
7.2.5.4.2 Place the tee piston in the tee position and adjust the height of the level bottle so that the liquid level of the measuring pipe is stable at the zero marking line. Then fix the position of the level cylinder, turn the three-way piston clockwise 135°, so that the reaction device is in an airtight state.
7.2.5.4.3 Read the temperature displayed by the thermometer in the water bath or water cooling jacket, convert it into Kelvin temperature, and take it as the temperature T of the initial state of the gas in the gas pipe.
7.2.5.5 Heating reaction
7.2.5.5.1 Remove the water bath and dry the outer wall of the conical bottle. Tilt the conical bottle together with the condensing tube to tip the weighing bottle and immerse the sample into the sodium hydroxide solution inside the conical bottle.
7.2.5.5.2 Place the electric stove plate under the conical bottle for electric heating. After the solution in the conical bottle boils, reduce the power of the electric furnace and keep it in a slightly boiling state. In this process, due to the production of a large amount of hydrogen, the liquid level of the sealing liquid in the measuring pipe will be sharply reduced, and attention should be paid to reducing the height of the level bottle in time, so that the level bottle and the measuring pipe (or gas storage cylinder) of the liquid level height difference is maintained within 50mm.
7.2.5.5.3 When the liquid level of the sealing liquid in the measuring pipe (or gas storage cylinder) is basically no longer changing, continue heating for 5min~10min, and then stop heating and remove the electric furnace pan. Generally, the total heating time is about 30min~80min, and the reaction can be complete.
7.2.5.6 Determination of final state parameters and apparent gas production
7.2.5.6.1 Immerse the conical bottle after heating reaction into the water bath again for cooling. During the cooling process, the height of the leveling bottle should be adjusted in time to keep the liquid level in the measuring pipe basically level with the liquid level of the leveling bottle.
7.2.5.6.2 When the water temperature in the water bath is consistent with the water temperature in the water cooling jacket, fix the position of the level bottle and continue to cool for about 5min to 10min. If the liquid level change of the measuring pipe does not exceed 0.2ml in the last 5 minutes, rotate the three-way piston 45 clockwise to make the reference tube connected with the atmosphere.
7.2.5.6.3 Adjust the height of the level bottle so that the liquid level in the measuring pipe and the reference pipe are on the same level line, fix the level bottle, and read the apparent gas production V from the liquid level of the measuring pipe, accurate to 0.2mL.
7.2.5.6.4 Read the temperature displayed by the thermometer in the water-cooled jacket and convert it into Kelvin temperature, which is used as the final state temperature T of the gas in the gas pipe.
7.2.5.6.5 Use a barometer to measure atmospheric pressure P accurately to 0.01kPa. It should be corrected according to the instruction manual of the instrument.
7.2.5.6.6 Turn the three-way piston to the three-way position, turn off the constant temperature water cooling device, and remove the conical bottle to end the test.
7.2.5.7 Determination of true gas generation
When the initial and final cooling water temperatures |T0-T1| are less than or equal to 1.0K, the apparent gas generation V1 is taken as the real gas generation V0. If the test facility cannot reach the condition |T0-T1|≤1.0K, the real gas generation V can be calculated according to formula (3) :
![]()
Formula:
V0- apparent gas production in milliliters (mL);
Vi- atmospheric pressure in the laboratory, in kilopascal (kPa);
T0,T1- The temperature of the water in the initial and final water bath or water cooling jacket, in Kelvin (K);
P0,P1 — temperature T. T, saturated vapor pressure of pure water, unit is kilopascal (kPa);
V0– the total effective volume between the conical bottle of the measuring device and the zero scale line of the measuring pipe, in milliliters (mL);
The volume of sodium hydroxide solution (7.2.2.1) added to the V2 conical bottle, in milliliters (mL);
V3 the volume of the sample, hydrochloric acid and deionized water in the sample, in milliliters (mL);
V4 The volume of the sodium hydroxide solution portion of the cylindrical scale, expressed in milliliters (mL).
7.2.6 Result calculation
Elemental silicon quantity is calculated by mass fraction w(Si), the value is expressed as %, calculated according to equation (4) :
![]()
Shizhong
P- Atmospheric pressure in the laboratory in kilopascals (kPa);
P1- Saturated vapor pressure of pure water at temperature T1, in kPa;
V- Actual gas production in milliliters (mL);
m The number of samples weighed, expressed in grams (g);
T1- Measure the temperature of the gas in the trachea at the final state, expressed in Kelvin (K);
0.001689- coefficient of hydrogen volume converted to elemental silicon mass.
7.2.7 Allowed difference
The difference between the two effective analysis values of the method should not be greater than the allowable difference specified in Table 3.
|
Table 3 Allowable difference of elemental silicon analytical value by gas volumetric method |
|
|
w(Si)/% |
Allowable error/% |
|
>0.5~≤5 |
0.10 |
|
>5~≤10 |
0.20 |
|
>10~≤99 |
0.50 |
8 Determination of silica
8.1 General Provisions
This method is not applicable to industrial silicon products containing silicon carbide.
Silica determination can be carried out in the following two ways:
a) Molybdenum blu-ray method (0.5%~10%)(8.2);
b) Weight-molybdenum blu-ray method (>10%)(8.3).
8.2 Molybdenum Blu-ray method (0.5%~10%)
8.2.1 Principles
After the sample was soaked in sodium hydroxide solution, the silica did not dissolve. After separating alkali insoluble matter, the residue was melted with mixed flux, and the amount of silica in the solution was determined by molybdenum blu-ray method.
8.2.2 Reagents
8.2.2.1 Mixed flux: Grind two parts anhydrous sodium carbonate with one part boric acid and mix well.
8.2.2.2 Ammonium molybdate solution (50g/L): Use after filtration.
8.2.2.3 Oxalic acid and sulfuric acid mixture: Weigh 25g oxalic acid, dissolve in 250ml sulfuric acid (1+8), dilute with water to 1000mL, and mix well.
8.2.2.4 Ammonium ferrous sulfate solution (50g/L): Weigh 50g ammonium ferrous sulfate and dissolve it in a certain amount of water, add 10mL sulfuric acid (1+1), and finally dilute it with water to 1000mL, mix it well, and filter it before use. Use time to prepare.
8.2.2.5 Hydrochloric acid (1+4).
8.2.2.6 Anhydrous ethanol.
8.2.2.7 Standard storage solution of silica (including SiO,0.5mg/ml): Weigh 0.1000g silica (purity 99.99%) which has been previously burned at 1000℃ for 2h and cooled to room temperature in a platinum crucible and add 2g~3g anhydrous sodium carbonate (reference reagent), cover the crucible cover and leave a gap, melt in 1000℃ high temperature furnace for 5min~10min, remove and cool. Place in a teflon beaker containing 100mL boiling water, heat at low temperature to leach the frit until the test liquid is clear, wash the crucible and cover with hot water, and cool to room temperature.Transfer 200mL into a volumetric bottle, dilute to scale with water, shake well, and store in a dry clean plastic bottle.
8.2.2.8 Silica standard storage solution (including Si0,50μg/mL): Transfer 25.00ml silica storage solution (8.2.2.7) to 250mL, dilute it with water to the scale, shake well, and store it in a dry clean plastic bottle.
8.2.3 Sample size
Weigh the sample about 0.2g, accurate to 0.1mg.
8.2.4 Measurement
8.2.4.1 Put the filter paper and residue (7.1.4.1) after leaching and filtering with sodium hydroxide solution (7.1.2.1) into a platinum crucible, dry and ash, add a mixed flux of 1g~2g, put into a high temperature furnace of 800℃~900℃, and melt at a temperature of 1000℃~1050℃ 5min~15min, remove and cool.
8.2.4.2 Add 100mL hot hydrochloric acid (8.1.2.5) into 200mL beaker, leach the melt at low temperature, cool it, transfer it to 200ml volumetric bottle, and dilute it with water to the scale. Shake well.
8.2.4.3 Remove 10.00mL and test solution (8.2.4.2)[When w(SiO,) in the sample is >5%, remove 5.00mL and test solution, add 5mL and test blank ]iquid] in 100mL volumetric bottle.
8.2.4.4 Add 10mL, anhydrous ethanol (8.2.2.6) and 5ml ammonium molybdate solution (8.2.2.2), shake well, and place at room temperature for 20min~30min(if the room temperature is lower than 15℃, place in a warm bath of about 30℃). 8.2.4.5 Add 25m, oxalate-sulfuric acid mixed solution (8.2.2.3), shake well, leave for 2min, add 5ml ammonium ferrous sulfate solution (8.2.2.4), dilute with water to the scale, shake well.
8.2.4.6 The absorbance of a suitable absorption dish (see Table 4) was measured at 690nm wavelength of the spectrophotometer with a blank test solution as a reference.
|
Table 4 Selection of absorption dishes according to silica content |
||
|
w(SiO₂)/% |
0.5~2.5 |
2.5~10 |
|
Absorption dish/mm |
10 |
5 |
|
Standard curve |
8.2.5 |
8.2.5 |
8.2.5 Drawing of standard curves Take 0mL, 1.00mL, 2.00mL, 4.00mL, 6.00mL, 8.00mL, 10.00ml silica standard solution (8.2.2.8) and put it into a group of 100ml volumetric bottles, add 2mL, Hydrochloric acid (8.2.2.5), add water to 20ml. Perform operations from 8.2.4.4 to 8.2.4.6. The absorbance was measured with reagent blank as reference, and the standard curve was drawn.
8.2.6 Calculation of analysis results
Silica is measured by mass fraction w(SiO,), the value is expressed as %, calculated according to equation (5) :
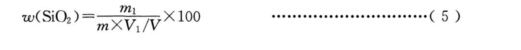
Formula:
m1- The amount of silica measured by a standard curve in grams (g);
m- The numerical value of the mass of the sample in grams (g);
V1- The value of the volume of the test solution, in milliliters (mL);
V- The value of the total volume of the test liquid in milliliters (mL).
8.3 Weight-Molybdenum Blu-ray method (>10%)
8.3.1 Principles
After the sample was soaked in sodium hydroxide solution, the silica did not dissolve. After separating the alkali insoluble matter, the residue is melted with a mixed flux, completely dried, condensed with polyoxyethylene to precipitate silicic acid, filtered and burned into silicon dioxide. Treat the burn with hydrofluoric acid. The quality difference before and after hydrofluoric acid treatment is the main amount of silica. The residual silica in the solution was determined by molybdenum blu-ray spectrophotometry after the residual was melted with hydrofluoric acid and leached with dilute hydrochloric acid.
8.3.2 Reagents
The sum of the two is the amount of silica in the sample.
8.3.2.1 Mixed flux: Grind two parts anhydrous sodium carbonate and one part boric acid and mix well.
8.3.2.2 Ammonium molybdate solution (50g/1): Use after filtration. 8.3.2.3 Oxalic acid and sulfuric acid mixture: Weigh 25g oxalic acid, dissolve with 250mL sulfuric acid (1+8), dilute with water to 1000mL, and mix well. 8.3.2.4 Ammonium ferrous sulfate solution (50g/L): Weigh 50g ammonium ferrous sulfate and dissolve it in a certain amount of water, add 10mL sulfuric acid (1+1), and finally dilute it with water to 1000mL, mix it well, and filter it before use. Use time to prepare.
8.3.2.5 p-nitrophenol (5g/L): Prepared with ethanol.
8.3.2.6 Sodium Hydroxide solution (200g/L): Store in plastic bottles.
8.3. 2.7 Hydrochloric acid (p=1.19g/mL).
8.3. 2.8 Sulfuric acid (p=1.42g/mL)
8.3.2.9 Hydrofluoric acid (p=1.13g/mL).
8.3.2.10 Hydrochloric Acid (1+4)
8.3.2.11 Hydrochloric acid (1+1),
8.3.2.12 Hydrochloric Acid (5+95)
Polyvinyl oxide (1g/1): Weigh 0.2g polyvinyl oxide and dissolve it in 200m water.
8.3. 2.13 Valid for two weeks.
8.3. 2.14 Silver nitrate :10g/L.
8.3.2.15 Standard storage solution of silica (including SiO,0.5mg/mL): Weigh 0.1000g silica (purity 99.99%) which has been burned at 1000℃ for 2h and cooled to room temperature in a platinum crucible, and add 2g~3g anhydrous sodium carbonate (standard)
8.3.2.16 Silica standard storage solution (including SiO,50μg/ml): Remove 25.00ml, silica storage solution (8.3.2.15) into 250mL volumetric bottle, dilute to scale with water, shake well, and store in dry clean plastic bottle.
8.3.2.17 Standard silica storage solution (including SiO,5g/mL): Transfer 25.00m silica storage solution (8.3.2.16) to 250ml, dilute it with water to the scale, shake well, and store it in a dry clean plastic bottle.
8.3.3 Sample size
Weigh the sample about 0.2g, accurate to 0.1mg.
8.3.4.1 The filter paper and residue (7.1.4.1) after being soaked and filtered by sodium hydroxide solution (7.1.2.1) are put into a platinum crucible, dried for ashing, added with a mixed flux of 1g~2g, put into a high temperature furnace of 800℃~900℃, and melt at a temperature of 1000℃~1050℃ 5min~15min, remove and cool.
8.3.4.2 Place 40ml of boiling, hydrochloric acid (8.3.2.11) and 50mL of water in a 200ml beaker. The melt is leached by heating. After cooling, add 10ml hydrochloric acid (8.3.2.7), 10mL, polyoxide ethylene (8.3.2.13), rinse the inner wall of the beaker and the glass rod with 10ml hot water, stir thoroughly for 1min~2min, and then rinse the inner wall of the beaker and the glass rod with 10ml hot water again Heat the hot plate at 50℃~70℃ for 10min~15min, so that the soluble salts are completely dissolved. Filter with slow filter paper. Filtrate is contained in 250ml volumetric bottle B. Transfer the precipitation from the beaker to the funnel with hot hydrochloric acid (8.3.2.12). Scrub the inner wall of the beaker and the glass rod with a small piece of filter paper and transfer them to the funnel. Wash in hot water until no chloride ions are present (check with silver nitrate solution (8.3.2.14)).
8.3.4.3 Place the precipitation together with filter paper (8.3.4.2) in a platinum crucible for drying and ashing. Put it in a high temperature furnace of 1000℃~1050℃ for 1h, take it out, cool it in a dryer to room temperature, and weigh it. This operation is repeated (15min each time) until the constant weight (m). 8.3.4.4 The precipitation after burning should be moistened with water, add 1 to 2 drops of sulfuric acid (8.3.2.8), 5m, hydrofluoric acid (8.3.2.9), evaporate at low temperature until white sulfuric acid smoke, take it off slightly cold, and add another 2ml,~3mL. Hydrofluoric acid (8.3.2.9), continue to evaporate until dry, at this time the temperature is slightly increased, carefully heated to remove the white sulfuric acid smoke, and then placed in a high temperature furnace,1000 ° C ~1050 ° C for 30 minutes, remove it, put it in a dryer, cool to room temperature, weigh. This operation is repeated (15min each time) until the constant weight (m).
8.3.4.5 Add about 1g of mixed flux (8.3.2.1) into the burned platinum crucible (8.3.4.4) and melt it in a high temperature furnace at 1000℃~1050℃ for 5min~15min. Remove and cool, add 5ml, leach with hydrochloric acid (8.3.2.11), combine into the volume bottle B to receive the original filtrate, dilute with water to the scale, and shake well.
8.3.4.6 Divide 10.00mL from volumetric bottle B, add 1 drop of p-nitrophenol (8.3.2.5) into 100mL plastic beaker, adjust to yellow with sodium hydroxide (8.3.2.6), and add 2mL hydrochloric acid (8.3.2.10). Transfer the solution into a volume bottle of 100ml.
8.3.4.7 Add 5ml of ammonium molybdate (8.3.2.2), shake well, and place at room temperature for 20min~30min(if the room temperature is lower than 15℃, do it in warm water at about 30℃). Add 25ml of oxalate-sulfuric acid mixture (8.3.2.3), shake well and leave for 2min. Add 5ml, ammonium ferrous sulfate solution (8.3.2.4), dilute with water to scale, shake well.
8.3.4.8 The absorbance of a 30mm absorber was measured at the wavelength of a spectrophotometer at 810nm with a blank test solution as the reference.
8.3.5 For the drawing of the working curve, the standard silica solution of 0mL, 1.00mL, 2.00mL, 4.00mL, 6.00mL, 8.00ml and 10.00mL (8.3.2.17) was removed from the burette with a set of 100ml, respectively. In a volumetric bottle, add 2ml hydrochloric acid (8.3.2.10) and water to about 20mL~30mL. For details, see 8.3.4.7 and 8.3.4.8. The absorbance was measured with reagent blank as reference, and the working curve was drawn.
8.3.6 Calculation of analysis results
Silica is measured by mass fraction w(SiO,), the value is expressed as %, calculated according to equation (6) :
![]()
Formula:
m- The numerical value of the mass of the sample in grams (g);
The value of the mass of precipitation and crucible before treatment with m1-hydrofluoric acid, in grams (g);
m2-hydrofluoric acid treated precipitation and crucible mass value, in grams (g);
m3- The amount of silica measured by a working curve in grams (g);
m4- The value, in grams (g), of the mass of silica for blank gravimetric tests; V– the value of the volume of the test liquid, in milliliters (ml);
V- The value of the total volume of the test solution in milliliters (mL).
8.4 Tolerance
The difference between the two effective analysis values of the method should not be greater than the allowable difference specified in Table 5.
